Task-specific (columns) recommendation matrix for the density-functional approximation level/basis set (rows) combinations and composite methods. Color codes for our recommendations, while the text in the fields states the most significant expected errors at the respective level. See the original article for a detailed discussion.

Timings of r²SCAN-3c compared to related (semi-empirical and full DFT) approaches for a ~150 atom system with ~7000 basis functions with def2-QZVP (21_AB of the S30L Benchmark set).
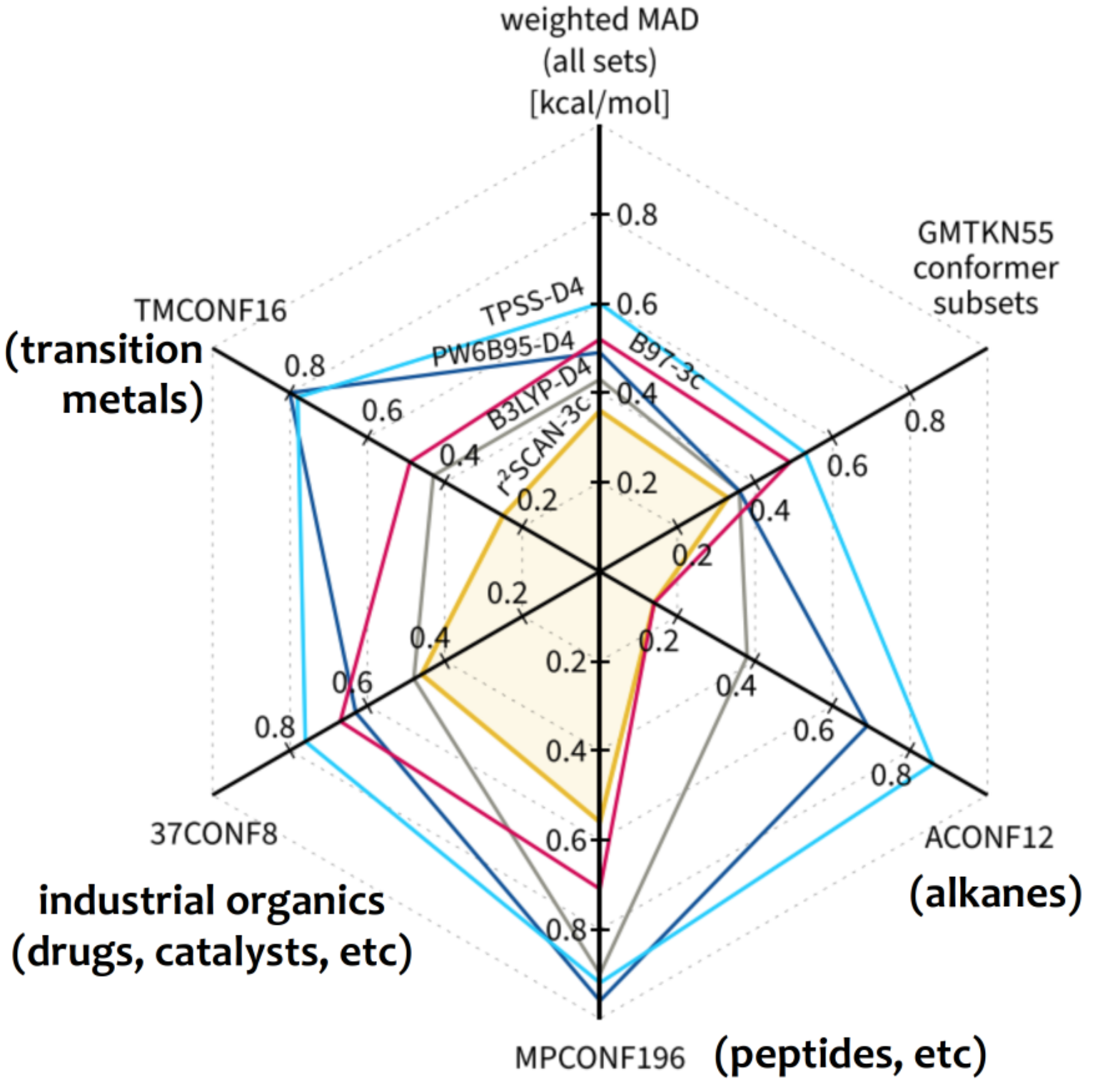
Accuracy of r²SCAN-3c for conformational energies of different classes of molecules compared to common DFT/QZ approaches from a Talk I gave in Hamburg @ DESY.
Density Functional Theory and Application
Work with Stefan Grimme, Georg Kresse, and Peter Schwerdtfeger
PAW Pseudopotentials and DFT-D3 Parameters for Superheavies - My first contact with DFT development was application-driven: The aim was to establish a consistent methodology for first-principles simulations of the Main Group superheavy elements Cn-Og (Z=112-118) to explore their bulk properties (see also Periodic Trends) together with Peter Schwerdtfeger. The first part of this methodology are Projector-Augmented Wave (PAW) pseudopotentials, the first of which I generated in cooperation with G. Kresse. These enable efficient plane-wave DFT simulations of the liquid and solid phases of the respective elements by taking care of the numerous (100) core electrons via an effective one-electron potential, which includes a large part of the relativistic effects (I also made non-relativistic PPs). The second part is an extension of the set of atomic parameters for DFT-D3 dispersion correction, which was done in cooperation with S. Grimme and S. Ehlert. These are particularly desirable for non-metallic elements, like relativistic Cn, Fl, and Og, where pure functionals like DFT/PBEsol provide the wrong asymptotic behavior (illustrated in Fig. 2 of this article on Og). The final methodology and some exemplary applications to cohesive energies and the adsorption on Au-surfaces can be found in this PCCP article “Exploring the chemical nature of super-heavy main-group elements through efficient plane-wave density-functional theory”, which made the cover page (left) and was designated as a “Hot Article”.
Refinement of DFT-D4 for Periodic Systems - After moving to Stefan Grimme’s group in Bonn, I joined forces with E. Caldeweyher, who was developing an extension of DFT-D4 for periodic systems. My experience with VASP—particularly for periodic systems, surfaces, and plane-wave DFT—proved invaluable during this work. Together, we benchmarked a comprehensive set of dispersion-corrected DFT methods for a wide range of applications, including the prediction of salt polarizabilities, the adsorption of polar molecules on various substrates, and the calculation of lattice and cohesive energies in both molecular crystals and metals. As expected, the extended D4 model outperformed D3, especially for highly coordinated Group 1–5 elements, where suitable reference data were lacking. Notably, both D3 and D4 derive polarizabilities and C6 coefficients via interpolation based on a fractional coordination number (CN), making the improved D4 approach particularly effective in these challenging cases.
r²SCAN-3c composite DFT Method - A recent project in collaboration with Stefan Grimme involved designing, implementing, and testing the r²SCAN-3c composite DFT method. It began with the publication of the re-regularized r²SCAN functional by Furness and coworkers, after Stefan recognized its potential for accurately handling conformational energies. While we were tailoring the components of this composite method—including the purpose-made mTZVPP basis set, specific parameters for DFT-D4, and a generalized gCP correction for basis-set superposition error—my primary responsibility was to ensure that the new method performed just as well for periodic solids and molecular crystals as it did for molecules. To this end, I benchmarked r²SCAN-3c against the X23, DMC8, and ICE10 test sets, as well as benzene adsorption on Cu, Ag, and Au, CO adsorption on MgO, and ethyne adsorption on NaCl. These results are showcased in the final article. Notably, the tests revealed that significantly increasing the three-body terms in the D4 model, along with carefully fine-tuning the charge scaling of D4 polarizabilities, provided clear benefits.
Beyond these developmental details, the resulting r²SCAN-3c demonstrates an impressive balance between computational cost and accuracy—so much so that I have already incorporated it into my day-to-day workflows. One of its most striking advantages is the ability to deliver “quick-but-not-dirty” conformational and reaction energies with accuracy rivaling hybrid-DFT/QZ-level methods, yet at two to three orders of magnitude lower computational cost. Another key strength lies in r²SCAN-3c’s improved short- and mid-range (mGGA) correlation, which surpasses that of its predecessor, SCAN. This improvement, in synergy with the long-range (semiclassical DFT-D4) dispersion correction, likely underpins the excellent performance of r²SCAN-3c for both adsorption energies (see Figure below) and intramolecular non-covalent interactions (conformational energies). Even years later, r²SCAN-3c continues to impress me with its robust performance for challenging systems such as lanthanide complexes and “Mindless” molecules—systems the method was never specifically trained on.
ω-dependency of the DFT-D damping function in RSHs - This project merges my work on DFT-D development with excited-state methods by investigating how variations in the range-separation parameter ω of range-separated hybrid (RSH) functionals affect dispersion-correction parameters. This issue is particularly relevant for functionals often used with optimal tuning, where the default DFT-D parameters might not remain suitable. Initial results show that PBE-derived RSHs (ωPBE, ωPBEh) and ωB97M-V retain robust performance over a wide ω-range, while BLYP/B88-based RSHs—especially LC-BLYP and CAM-QTP01—exhibit anomalies even at moderate ω-values, suggesting a deeper problem in the B88 exchange functional or μB88 range-separated GGA exchange. These findings have been compiled into a manuscript currently under review.
Dynamic Radii Adjustment for COntinuum Solvation - Just like the classic “spherical cows in a vacuum” joke highlights oversimplifications in science, many computational chemistry methods treat molecules as if they have fixed atomic sizes—ignoring how the solvent environment really affects molecular shape. Enter DRACO (Dynamic Radii Adjustment for Continuum Solvation), a method I co-developed to break free of static, element-based radii in polarizable continuum models (PCMs). DRACO assigns each atom a size tailored to its local environment, using low-cost properties like approximate atomic charges and fractional coordination numbers (akin to D4’s approach). This clever tweak significantly boosts accuracy for challenging cases such as charged species—without ballooning computation time. During model development, we even discovered surprising trends (like shrinking negatively charged atoms) that revealed deeper insights into solvation physics. DRACO is now freely available in FACCTS ORCA6 and in TURBOMOLE, so you can easily incorporate it into your own workflows.
Experimental and calculated adsorption energies for benzene on the coinage metals, Au, Ag, and Cu, as well as CO on MgO and ethyne on NaCl. Calculations for r²SCAN-3c, PBE-D4, M06L-D4, and BLYP-D4 employ the same mTZVPP basis set as r²SCAN-3c, while B97-3c uses the slightly smaller def2-mTZVP basis set. SCAN-D4 and SCAN-rVV10 have been calculated with VASP using a plane-wave cutoff of 700 eV. If available, contributions from the DFT part (blue), the dispersion correction (orange), and the gCP term (yellow) are given separately. The increase in the binding energy on Au due to spin–orbit coupling has been calculated with SCAN in VASP and added to the other results (bright blue). The MAD is given in green.
